Code
library(CoRC)
# Data wrangling
library(dplyr)
library(readr)
# Table visualisation
library(scales)
library(flextable)
# Plot visualisation
library(ggplot2)
library(latex2exp)
library(cowplot)
source("../../scripts/utils.R")The list of packages required for reproducing the analyses on this script are listed in Listing 1. Besides, the downloadable R script file utils stores two helper functions:
get_legend_temp is a temporary fix to retrieve the legend of any ggplot2 or grob object (used for further customisation of a panel of subplots). For further details on this custom fix, report to Open Cowplot Issue for shared legend feature.colourer is a custom styling function displaying all positive values as red, and negative ones as blue (used with flextable).library(CoRC)
# Data wrangling
library(dplyr)
library(readr)
# Table visualisation
library(scales)
library(flextable)
# Plot visualisation
library(ggplot2)
library(latex2exp)
library(cowplot)
source("../../scripts/utils.R")To run an experimental design, we need:
Note that Lo et al. (2016) did not provide unfortunately the individual cytokine concentrations, preventing from reproducing the differential analyses and quite hampering the validity of Parameter estimation Task.
# Load Healthy ODE model along with the parameter constants
healthy_model <- loadModel("../../models/team_2016_final_model_lo2016_2025_05_13.cps")
healthy_steaty_state <- runSteadyState(
calculate_jacobian = FALSE,
model = healthy_model
)$species |>
dplyr::select(name, concentrationH = concentration)
# Load experimental datasets providing real-life concentrations of cytokines
cluster_type <- c("Type 1", "Type 2", "Type 3", "Type 4")
data_experiment <- readr::read_csv("../../data/IBD DGEA.csv", show_col_types = FALSE) |>
tidyr::pivot_longer(!cluster, names_to = "name", values_to = "concentration") |>
dplyr::left_join(healthy_steaty_state, by = "name") |>
mutate(
concentration = if_else(!is.na(concentrationH),
concentrationH * (1 + concentration / 100), concentration
),
concentrationH = NULL
) |>
tidyr::pivot_wider(names_from = name, values_from = concentration) |>
split(f = cluster_type)Types of disease | Species | Species' concentration at steady-state | Concentrations in IBD clusters | fold change |
|---|---|---|---|---|
type 1 | I6 | 1.46e-06 | 1.02e-07 | -93.0% |
type 1 | I10 | 3.01e-08 | 1.48e-08 | -51.0% |
type 1 | Ia | 2.72e-05 | 1.88e-05 | -31.0% |
type 1 | Ib* | 1.64e-13 | 7.23e-14 | -56.0% |
type 1 | Ig* | 2.01e-07 | 2.01e-09 | -99.0% |
type 2 | I6 | 1.46e-06 | 3.78e-07 | -74.0% |
type 2 | I10 | 3.01e-08 | 2.56e-08 | -15.0% |
type 2 | Ia | 2.72e-05 | 1.88e-05 | -31.0% |
type 2 | Ib* | 1.64e-13 | 1.54e-13 | -6.0% |
type 2 | Ig* | 2.01e-07 | 1.61e-08 | -92.0% |
type 3 | I6 | 1.46e-06 | 1.96e-06 | 35.0% |
type 3 | I10 | 3.01e-08 | 4.34e-08 | 44.0% |
type 3 | Ia | 2.72e-05 | 4.00e-05 | 47.0% |
type 3 | Ib* | 1.64e-13 | 1.95e-13 | 19.0% |
type 3 | Ig* | 2.01e-07 | 1.17e-06 | 481.0% |
type 4 | I6 | 1.46e-06 | 2.47e-07 | -83.0% |
type 4 | I10 | 3.01e-08 | 1.99e-08 | -34.0% |
type 4 | Ia | 2.72e-05 | 1.49e-05 | -45.0% |
type 4 | Ib* | 1.64e-13 | 1.31e-13 | -20.0% |
type 4 | Ig* | 2.01e-07 | 1.00e-08 | -95.0% |
*These quantities have not been measured, but rather estimated from the model. | ||||
Species is the cytokines’ name, while cluster indicator is returned by Types of disease. Note that we have not reported species concentrations of \(T-Bet\), \(Gata3\), \(ROR \gamma t\), and \(Foxp3\) as not measured in the ODE model.
# define larger lower and upper bounds
free_parameters_key <- getParameters(c(
"(Prod of Ig from T1).k1",
"(Prod of Ia from T1).k1",
"(Prod of Ib from Tr).k1",
"(Prod of I10 from Tr).v10r",
"(Induction of T1 from M).s12",
"(Induction of T2).s4",
"(Induction of T17).s21",
"(Induction of T17).s6",
"(Induction of Tr).sb",
"(Induction of Tr).s10"
), model = healthy_model)$key
fit_parameters <- purrr::map(free_parameters_key, function(param) {
val <- getParameters(param)$value
defineParameterEstimationParameter(
ref = parameter(param, "Value"),
start_value = val,
lower_bound = 1e-15,
upper_bound = 1e4
)
})
mapping_present <- getSpeciesReferences(
key = c("I6", "I10", "Ia", "Ib", "Ig"),
model = healthy_model
) |> pull(concentration)
fit_experiments <- purrr::map(data_experiment, ~ defineExperiments(
experiment_type = "steady_state",
data = .x,
types = c(
"ignore", "dependent", "dependent", "dependent",
"ignore", "ignore", "ignore", "ignore", "ignore", "ignore"
),
mappings = c(NA, mapping_present, rep(NA, 4)),
weight_method = "mean_square"
))free_parameters_key <- getParameters(c("(Prod of Ig from T1).k1",
"(Prod of Ia from T1).k1",
"(Prod of Ib from Tr).k1",
"(Prod of I10 from Tr).v10r",
"(Induction of T1 from M).s12",
"(Induction of T2).s4",
"(Induction of T17).s21",
"(Induction of T17).s6",
"(Induction of Tr).sb",
"(Induction of Tr).s10"), model = healthy_model)$key
fit_parameters <- purrr::map(free_parameters_key, function(param) {
val <- getParameters(param)$value
defineParameterEstimationParameter(
ref = parameter(param, "Value"),
start_value = val,
lower_bound = 1e-15,
upper_bound = 1e4)
})
mapping_present <- getSpeciesReferences(
key = c("I6", "I10", "Ia", "Ib", "Ig"),
model = healthy_model
) |> pull(concentration)
fit_experiments <- purrr::map(data_experiment, ~ defineExperiments(
experiment_type = "steady_state",
data = .x,
types = c(
"ignore", "dependent", "dependent", "dependent",
"ignore", "ignore", "ignore", "ignore", "ignore", "ignore"),
mappings = c(NA, mapping_present, rep(NA, 4)),
weight_method = "mean_square"
))steady-state, or time_course when several time points have been considered in the design), the measured concentrations diverging from the average concentrations in healthy patients with data, define each of the measured variables (constant with independent, dependent if used for tailoring the parameter estimates, or ignore if not quantified in the ODE model). We had only the averaged expression per patient subgroup To better capture within-patient variability, it would have been valuable to provide the individual cytokine profiles, and perform a multiple least-squares regression, aka MMR task.
mappings guarantees the proper pairing between COPASI variables and string values of column names2.
weight_method] coupled with weights describe for each variable the assumed mean-variation trend. Report to COPASI Manual, section Experimental Data, for further details.
Finally, once the experimental design and optimisation criteria have been configured independently for each free parameter and patient subgroup, we have to run the parameter estimation itself:
absolute_parameter_per_cluster <- purrr::map(fit_experiments, function(cluster) {
# CoRC update imposes to restart from a clean model for parameter estimation
CoRC::clearParameterEstimationParameters(model = healthy_model)
CoRC::clearExperiments(model = healthy_model)
return(runParameterEstimation(
parameters = fit_parameters,
experiments = cluster,
method = list(
method = "LevenbergMarquardt",
log_verbosity = 0,
iteration_limit = 200,
tolerance = 1e-5
),
update_model = FALSE,
randomize_start_values = FALSE,
create_parameter_sets = TRUE,
calculate_statistics = FALSE,
model = healthy_model
)$parameters)
})
# Make results directly comparable with Table 8 by switching from absolute to relative ratios
relative_parameter_per_cluster <- absolute_parameter_per_cluster |>
purrr::list_rbind(names_to = "cluster") |>
dplyr::select(cluster, parameter, start_value, value) |>
mutate (relative_concentration = 100*(value - start_value)/start_value,
start_value = NULL, value = NULL) |>
tidyr::pivot_wider(names_from = parameter, values_from = relative_concentration) absolute_parameter_per_cluster <- purrr::map(fit_experiments, function(cluster) {
CoRC::clearParameterEstimationParameters(model = healthy_model)
CoRC::clearExperiments(model = healthy_model)
return(runParameterEstimation(
parameters = fit_parameters,
experiments = cluster,
method = list(method = "LevenbergMarquardt",
iteration_limit = 500,
tolerance = 1e-6),
update_model = FALSE,
randomize_start_values = FALSE,
create_parameter_sets = TRUE,
calculate_statistics = FALSE,
model = healthy_model
)$parameters)
})clearParameterEstimationParameters and clearExperiments guarantee a fresh start for parameter estimation for each scenario.
runParameterEstimation, of the pipeline for estimating varying constants across distinct biological conditions.
iteration_limit determines the maximum number of iterations the algorithm shall perform, while tolerance is the numerical precision to reach between two consecutive iterations.
Consider a multivariate function \(f(\mathbf{x})\) that we want to maximize with respect to \(\mathbf{x}\) (for instance, we may want to minimise the mean-squared error between estimated steady-state concentrations and ones measured in each patient subgroup). Standard unconstrained optimization approaches include Gradient Descent (see Equation 1), Newton-Raphson (see Equation 2) and Levenbergh-Marquart (see Equation 3):
The Steepest Descent Approach, where the method updates \(\mathbf{x}\) in the direction of the gradient (steepest slope):
\[ \mathbf{x}_{k+1} = \mathbf{x}_k + \alpha_k \nabla f(\mathbf{x}_k) \tag{1}\]
where:
The Newton-Raphson Approach is a second-order method, requiring the computation of the Hessian matrix:
\[ \mathbf{x}_{k+1} = \mathbf{x}_k + \mathbf{H}^{-1}(\mathbf{x}_k) \nabla f(\mathbf{x}_k) \tag{2}\]
where:
On the other hand, the Newton method converges quadratically towards the minimum in its vicinity. However, it’s really sensible to the initialisation, and may not converge at all if it is far away from it.
The Levenberg-Marquardt Descent Approach (Equation 3):
\[ \mathbf{x}_{k+1} = \mathbf{x}_k + \left( \mathbf{H}(\mathbf{x}_k) + \lambda \mathbf{I} \right)^{-1} \nabla f(\mathbf{x}_k) \tag{3}\]
where \(\lambda\) is a dampening hyper-parameter controlling the trade-off between gradient descent, when \(\lambda \to \infty\), and Newton’s method \(\lambda \to 0\).
Our estimations are compared with Table 8, (Lo et al. 2016, 13) variations in Table 2.
Of note, all tables in the original paper were provided as screenshots and images, rather than CSV files, preventing straightforward reproducibility of the results.
# flextable formatting and variables
variable_labels <- list(
cluster = "Type of diseases",
`(Prod of Ig from T1).k1` = "coefficient of IFN-gamma by Th1",
`(Prod of Ia from T1).k1` = "coefficient of TNF-alpha by Th1",
`(Prod of Ib from Tr).k1` = "coefficient of TGF-beta by Treg",
`(Prod of I10 from Tr).v10r` = "coefficient of IL-10 by Treg",
`(Induction of T1 from M).s12` = "activation of Th1 cells",
`(Induction of T2).s4` = "activation of Th2 cells",
`(Induction of T17).s21` = "activation of Th17 cells",
`(Induction of T17).s6` = "activation of Th17 cells",
`(Induction of Tr).sb` = "activation of Treg cells",
`(Induction of Tr).s10` = "activation of Treg cells"
)
header_row <- c(
"\\text{Type of diseases}",
"\\nu_{\\gamma_1}",
"\\nu_{\\alpha_1}",
"\\nu_{\\beta_{\\rho}}",
"\\nu_{10_{\\rho}}",
"\\sigma_{12}",
"\\sigma_{4}",
"\\sigma_{21}",
"\\sigma_{6}",
"\\sigma_{\\beta}",
"\\sigma_{10}") |>
as_equation() |>
as_paragraph()
# flextable generation
relative_parameter_variations_flextable <- flextable(relative_parameter_per_cluster) |>
# Add gradient colouring based on values
bg(
j = setdiff(names(variable_labels), "cluster"),
bg = colourer) |>
# Format numbers (optional for consistent display)
colformat_double(j = setdiff(names(variable_labels), "cluster"),
suffix = "%",
big.mark = ",",
digits = 1) |>
append_chunks(j = "cluster",
# what to append
as_equation(c("\\text{Th1}\\uparrow \\text{Th2}\\downarrow",
"\\text{Th1}\\downarrow \\text{Th2}\\uparrow",
"\\text{Th1}\\uparrow \\text{Th2}\\uparrow",
"\\text{Th1}\\downarrow \\text{Th2}\\downarrow"))
) |>
# Rename columns using LaTeX-like notation
set_header_labels(values = variable_labels
) |>
add_header_row(values = header_row,
colwidths = rep(1, 11), top = FALSE
) |>
align(align = "center", part = "all") |>
set_caption("Table 8: Parameter variations in different types of diseases.
In blue, disease subtype induces shrinkage of the coefficients,
while red values correspond to increased parameter uptakes. ") |>
merge_at(part = "head", i = 1:2, j = 1)
relative_parameter_variations_flextableType of diseases | coefficient of IFN-gamma by Th1 | coefficient of TNF-alpha by Th1 | coefficient of TGF-beta by Treg | coefficient of IL-10 by Treg | activation of Th1 cells | activation of Th2 cells | activation of Th17 cells | activation of Th17 cells | activation of Treg cells | activation of Treg cells |
|---|---|---|---|---|---|---|---|---|---|---|
NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
Type 1NA | 64.3% | -2.6% | -18.7% | 82.9% | -2.0% | 15.7% | -100.0% | -100.0% | 268.7% | 333.3% |
Type 2NA | -3.5% | -2.7% | 74.0% | -100.0% | -2.1% | 16.2% | -100.0% | -100.0% | 264.4% | 327.8% |
Type 3NA | 517,571.6% | 17.7% | 501,029,061,480.2% | 274,184,766.1% | 6.9% | -91.7% | -100.0% | 6,303.3% | -100.0% | -100.0% |
Type 4NA | 62,894.1% | -4.0% | -96.2% | 315.2% | -3.1% | 24.1% | -100.0% | -100.0% | 378.0% | 468.7% |
One of the major advantages of flextable lies in its ability to save the generated flextable object in a variety of formats, including docx, and html, as illustrated in Listing 3 (or even several ones simultaneously).
save_as_html(relative_parameter_variations_flextable,
path = "../../results/Table8.html",
title = "Table 8: Parameter variations per disease subtype."
)
save_as_docx(relative_parameter_variations_flextable,
path = "../../results/Table8.docx",
title = "Table 8: Parameter variations per disease subtype."
)The objective of this section is to predict the efficiency of a novel anti-TNF-\(\alpha\) treatment, assuming that the blocking treatment is equivalent to force the concentration of TNF-\(\alpha\) to remain zero, and calculate the change of other variables at the steady-state concentrations.
Second objective is to determine the efficiency of the treatment conditioned to the patient endotype determined using patient stratification approaches.
Since the results of parameter estimation reported in Section 1.3 differ significantly from the results reported in the original paper, Table 8, Lo et al. (2016), pp. 13, we directly retrieve in that section the original estimates from Lo et al. (2016).
## Prepare parameter variations for each of the four patient subtypes ----
healthy_model <- suppressWarnings(loadModel("../../models/team_2016_final_model_lo2016_2025_05_13.cps"))
# retrieve parameter mapping
parameters_per_clusters <- getParameters(c("(Prod of Ig from T1).k1",
"(Prod of Ia from T1).k1",
"(Prod of Ib from Tr).k1",
"(Prod of I10 from Tr).v10r",
"(Induction of T1 from M).s12",
"(Induction of T2).s4",
"(Induction of T17).s21",
"(Induction of T17).s6",
"(Induction of Tr).sb",
"(Induction of Tr).s10"), model = healthy_model)
# retrieve parameter variations specific to each disease subtype ...
patient_group <- c("Type1", "Type2", "Type3", "Type4")
parameters_per_clusters <- parameters_per_clusters |>
dplyr::inner_join(readr::read_csv("../../data/IBD subgroups.csv", show_col_types = FALSE), by = "key") |>
rename(healthy = value) |>
dplyr::select(-mapping) |>
# ... and convert relative ratios to absolute
dplyr::mutate(across(all_of(patient_group), \(x) healthy * (1 + x/100)))
# run healthy steady state
healthy_steaty_state <- runSteadyState(
calculate_jacobian = FALSE,
model = healthy_model)$species |>
dplyr::select(name, concentrationH = concentration) |>
filter (name %in% c("I6", "I10", "Ia", "T1", "T2","T17", "Tr", "Ig", "Ib"))steady_states_per_cluster <- purrr::map(patient_group, function(cluster) {
# without anti-TNF alpha treatment
disease_model_without_treatment <- healthy_model |> saveModelToString() |> loadModelFromString()
setParameters(model = disease_model_without_treatment,
key = parameters_per_clusters$key,
value = parameters_per_clusters[[cluster]])
without_treatment_steady_state <- runSteadyState(
calculate_jacobian = FALSE,
model = disease_model_without_treatment)$species |>
dplyr::select(name, concentration) |>
inner_join(healthy_steaty_state, by = "name") |>
mutate (concentration = 100* (concentration - concentrationH) / concentrationH) |>
dplyr::select(-concentrationH) |>
mutate(cluster = cluster, category = "untreated")
# with anti-TNF alpha treatment
disease_model_with_treatment <- suppressWarnings(disease_model_without_treatment |>
saveModelToString() |>
loadModelFromString())
setSpecies(key = "Ia{compartment}",
initial_concentration = 0,
type ="fixed",
model = disease_model_with_treatment)
with_treatment_steady_state <- runSteadyState(
calculate_jacobian = FALSE,
model = disease_model_with_treatment)$species |>
dplyr::select(name, concentration) |>
inner_join(healthy_steaty_state, by = "name") |>
mutate (concentration = 100* (concentration - concentrationH) / concentrationH) |>
dplyr::select(-concentrationH) |>
tibble::add_row(name = "Ia", concentration = 0) |>
mutate(cluster = cluster, category = "treated")
return (rbind(without_treatment_steady_state, with_treatment_steady_state))
}) |> dplyr::bind_rows()fixed and null by setting the initial concentration level to \(0\). Both parameters are required to ensure absence of TNF-\(\alpha\).
dplyr::bind_rows() merges a list of data.frame into a single one.
We have reproduced Fig.3 from Lo et al. (2016), pp. 13, in Figure 1, using a combination of ggplot2for individual barplot representations and cowplot for individual ‘ggplot’ concatenations.
# Reproduce boxplots of Figure 3, pre and post-treatment ----
# Format dataset for plotting
steady_states_per_cluster_plot <- steady_states_per_cluster |>
mutate (name = factor (name,
levels = c("I6", "I10", "Ia", "T1", "T2",
"T17", "Tr", "Ig", "Ib"),
labels = c("IL-6", "IL-10", "TNF-alpha", "Th1", "Th2",
"Th17", "Treg", "IFN-gamma", "TFG-beta"),
ordered = TRUE),
category = factor(category,
levels = c("untreated", "treated"), ordered = TRUE))
cluster_labels <- c("Type 1: Th1\\uparrow Th2\\downarrow",
"Type 2: Th1\\downarrow Th2\\uparrow",
"Type 3: Th1\\uparrow Th2\\uparrow",
"Type 4: Th1\\downarrow Th2\\downarrow")
names(cluster_labels) <- c("Type1", "Type2", "Type3", "Type4")
### Retrieve legend ----
global_plot <- ggplot(data = steady_states_per_cluster_plot,
mapping = aes(x = name, y = concentration, fill = category)) +
geom_col(position = "dodge") +
scale_fill_manual(name ="Treatment", values=c("blue", "red")) +
guides(color = guide_legend(nrow = 1)) +
theme(legend.position = "bottom")
### Generate bar plot for each disease subtype ----
list_plots <- purrr::imap(cluster_labels, ~ ggplot(data = steady_states_per_cluster_plot |> filter(cluster==.y),
mapping = aes(x = name, y = concentration, fill = category)) +
geom_col(position = "dodge")+
labs(x = NULL, y = NULL) +
scale_fill_manual(name ="Treatment", values=c("blue", "red")) +
theme_minimal() +
theme(panel.grid.major = element_blank(),
legend.position = "none", plot.title = element_text(hjust = 0.5)) +
ggtitle(TeX(.x)))
### Combine barplots into a 2x2 grid ----
plot_grid_combined <- cowplot::plot_grid(plotlist = list_plots,
ncol = 2, align = "hv", axis = "tblr")
x_label <- ggdraw() + draw_label("Cell Species", x = 0.5, y = 0.5)
y_label <- ggdraw() + draw_label("ODE Parameters", x = 0.5, y = 0.5, angle = 90)
# Add global y label
plot_grid_combined <- cowplot::plot_grid(y_label,
plot_grid_combined,
nrow = 1,
rel_widths = c(0.1, 1) # Adjust width for the legend
)
legend_plot <- get_legend_temp(global_plot)
# Add global x label
plot_grid_combined <- cowplot::plot_grid(plot_grid_combined,
x_label,
legend_plot,
nrow = 2, ncol =1,
rel_heights = c(1, 0.1) # Adjust width for the legend
)
plot_grid_combined <- cowplot::plot_grid(
plot_grid_combined,
legend_plot,
nrow = 2,
rel_heights = c(1, 0.1) # Adjust width for the legend
)
plot_grid_combined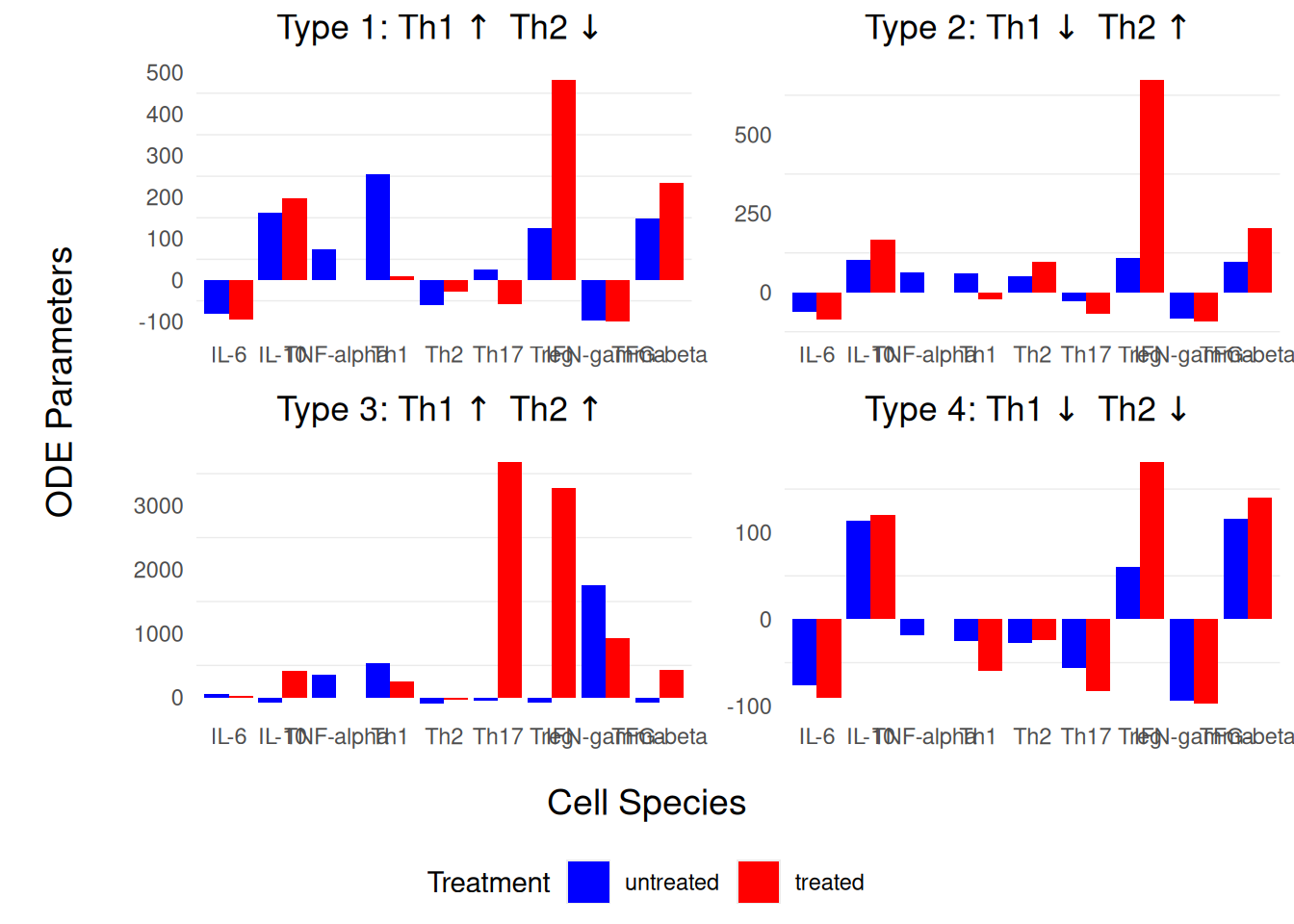
note the final split() instruction: indeed, the expected input is a list of experiences, each element of the list matching a subgroup of patients with their own set of free parameters↩︎
In contrast with documentation, this step seems mandatory, as the output returned by `` has a placeholder typical syntax.↩︎